This AI Startup Joins Forces With NVIDIA's BioNeMo to Boost Drug Discovery
Receptor.AI, a London-based drug discovery company and NVIDIA Inception member, has successfully integrated NVIDIA BioNeMo cloud APIs with their end-to-end Computer Assisted Drug Discovery (CADD) platform. In doing so, the company has achieved significant performance improvements and cost savings by shifting virtual screening, ADMET assessment and ligand pose prediction tasks from traditional CPU-based processing to accelerated computing on the NVIDIA platform.
Receptor.AI specializes in designing highly efficient therapeutics with an emphasis on selectivity and targeting protein-protein interactions (PPIs) using an in-house hybrid intelligence drug discovery platform. Usage of BioNeMo allowed the company to get access to the latest deep learning techniques backed up by the power of the full stack of NVIDIA hardware and software accelerations.
Receptor.AI demonstrated the power of their platform integrated with BioNeMo in a case study involving a human fatty acid desaturase 1 (FADS1) - an enzyme involved in the biosynthesis of polyunsaturated fatty acids that plays an important role in cancer genesis and biology. For the selectivity assessment against FADS1, three homologous fatty acid desaturase domains FADS2, FADS3, FADS6 were used as potential off-targets.
A chemical library consisting of ~4.3 billion small molecule compounds generated by MegaMolBART AI generator was used for virtual screening. 25 known FADS1 ligands were mixed into this generated chemical space. This allowed assessing the performance of Receptor.ai’s AI workflow in prioritizing known binder among other compounds.
Figure 1 outlines the scheme of the Virtual Screening experiment. The primary selection used the DTI and selectivity models, followed by the ADME-Tox assessment. DTI rank was used for proteome-wide selectivity assessment. BioNemo DiffDock AI docking was used to obtain the binding poses of shortlisted compounds. In the final stage, the Receptor.AI consensus function was used to rank the top candidate molecules.
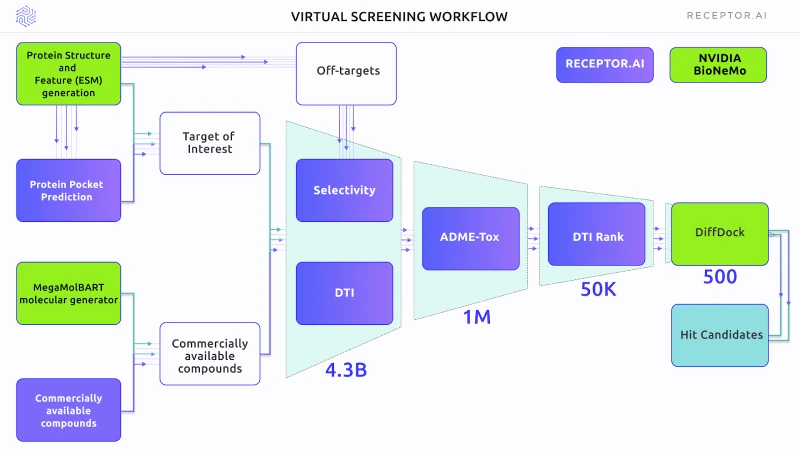
Figure 1. The general scheme of the experiment, including both BioNeMo (green blocks) and Receptor.AI (blue blocks) technology
The FADS1 protein structure was recomputed using BioNeMo AlphaFold. The ligand binding pocket was determined by the PUResNet AI model on a NVIDIA Tesla T4 GPU.
The Receptor.AI hit identification pipeline was used to narrow the chemical space from ~4.3 billion compounds to the 500 most promising candidates. The usage of Nvidia GPUs in the screening process provided speed-ups from 1.1x to 11.3x, depending on the task. The top 500 compounds were then docked blindly to the FADS1 without a priori information about the location of the binding pocket using the BioNeMo cloud API for DiffDock.
Receptor.AI has successfully identified several hit compounds from the known FADS1 ligands, as well as novel compounds with higher predicted affinity to FADS1, compared to the known ligands. Out of the 25 known FADS1 ligands, four were detected in the top 10 hit candidates. Almost half of all known ligands (12 of 25) were found in the top 1K. This indicates the utility of the proposed pipeline in discovering prospective hit compounds with novel molecular scaffolds. The predicted ADME-Tox parameters and the docking poses of the top three hit candidates are shown in Figure 2.
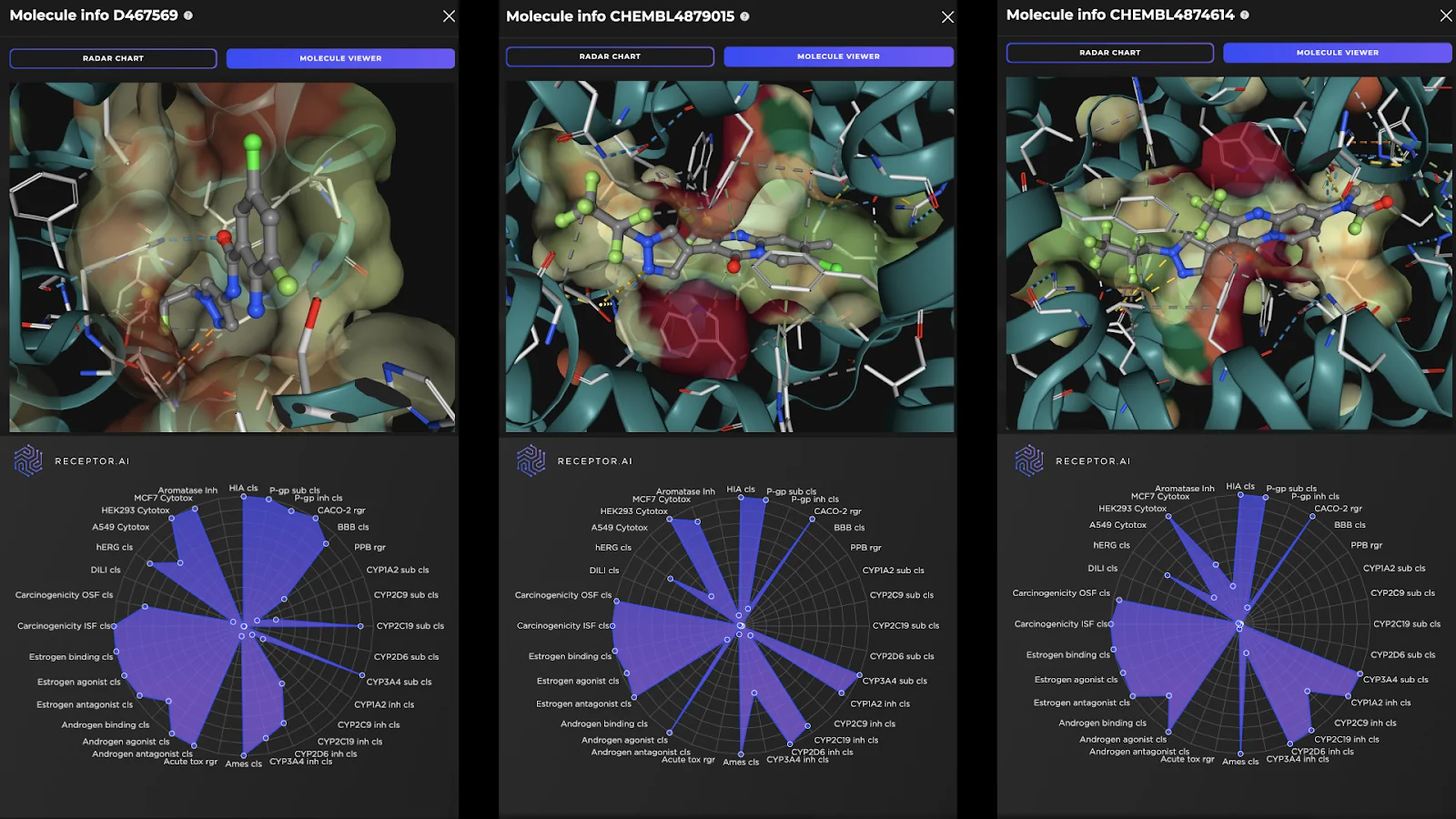
Figure 2. Docking poses (top panels) and the radar charts of ADME-Tox parameters (bottom panels) for the top three predicted FADS1 hit candidates
A significant finding for Receptor.ai in this case study was the cost-saving aspect of integrating accelerated computing into the virtual screening pipeline. This resulted in an overall 49% drop in the final cost, from $0.43 to $0.22 per instance-hour.
The next steps of this exciting collaboration will involve the integration of BioNeMo Framework to train Receptor.AI models with proprietary data and Receptor.AI advanced selectivity workflow. We aim to produce the ensembles of representative protein conformations by running molecular dynamics simulations on NVIDIA high-performance, accelerated workstations and cloud instances.
Moreover the ArtiDock 2.0, a flagship ligand pose prediction model from Receptor.AI, which outperforms all available physics-based and AI docking solutions in term of accuracy and speed, will soon be available through the BioNeMo API, which will push the AI-based docking solutions to the next level taking into account computational capabilities of Nvidia platform.
Further development of this technology stack will target creating high-performance AI-based drug discovery platforms for a wide range of biotech and pharma companies.
Comments:
There are no comments yet. You can be the first.
Leave a Reply cancel reply
Your email address will not be published. Required fields are marked *