Recursion Unveils New Foundation Model for Molecular Property Prediction
Recursion researchers have introduced MolE, a new model aimed at improving predictions of chemical and biological properties of molecules by leveraging molecular graphs and a unique pretraining approach. MolE builds on a foundation model concept, utilizing a large volume of unlabeled data to learn molecular patterns and a smaller labeled dataset to refine its predictions.
Capturing a molecule’s structure in a machine-readable form without losing essential information is complex. While many models use SMILES, a character-based molecular representation, this approach is limited by the lack of a unique format for each molecule, especially for larger structures. Instead of character sequences, MolE represents molecules as graphs, where atoms and bonds form structured connections that capture each molecule’s spatial arrangement—a critical factor in understanding molecular behavior. With this graph-based setup, MolE can learn from a vast volume of unlabeled data, reducing dependency on experimental datasets.
See also: Companies Pioneering AI Foundation Models in Pharma and Biotech
MolE’s Architecture
MolE is built on a variant of the transformer architecture called DeBERTa, incorporating a “disentangled attention” mechanism. In DeBERTa, attention weights take into account both the content of each token (representing atoms or bonds) and their relative positions within the molecular graph. This setup allows MolE to preserve spatial information about atomic arrangement, which is essential for accurate predictions of molecular behavior. In the paper, it's noted that this approach significantly improves MolE’s performance over other transformer models without positional embeddings.
MolE’s training approach is divided into two phases:
-
Self-Supervised Pretraining: MolE was first pre-trained in a self-supervised manner on a dataset of approximately 842 million molecules, where it learned to predict atom environments based on partial information. This step allows MolE to understand fundamental molecular structures without relying on labeled data.
-
Supervised Pretraining: MolE was then further trained in a supervised manner using a smaller labeled dataset of about 456,000 molecules, extracted from a subset of ChEMBL with labels spanning 1,310 tasks. This supervised phase fine-tuned the model’s understanding of chemical and biological properties, improving its predictive capabilities for real-world applications.
MolE was evaluated on the Therapeutic Data Commons (TDC) benchmark, which includes tasks like predicting absorption, distribution, metabolism, excretion, and toxicity (ADMET). MolE achieved top results on 10 of the 22 ADMET tasks, while the next best model, ZairaChem, led in only 5.
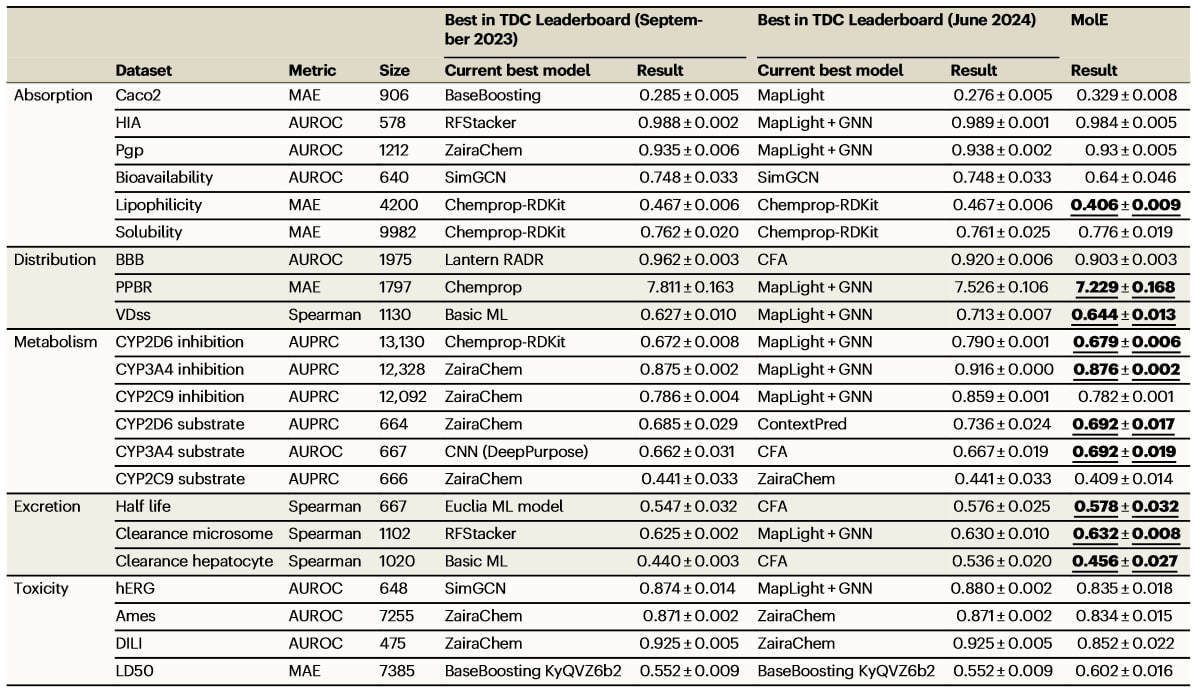
From paper: Comparison between the best models reported in the Therapeutic Data Commons (TDC) leaderboard (as of September 2023) and finetuned MolE
MolE shows that—by training on molecular graphs—transformer-based models can work quite effectively for chemistry applications, offering a new tool for predicting molecular properties with fewer data requirements.
For more details, read the full study in Nature Communications – DOI: 10.1038/s41467-024-53751-y.
Topics: Tools & Methods